If you want to get help directly related to a page/query, use the Info Pages / Medium Info Pages. Direct links are available at each (Query) Page.
If you are completely lost, here is link to a short description of what CSB.DB is and is not.
Enter this page.
Profiles: Single Metabolite Profil Search (sMPQ) 
Basic: The profiles can be queried by a particular compound name and allows searching for the co-responding compound levels within a particular experiment. Various filter options are available to restrict computation to high-quality mass traces / mass trace entries by using the default or user modified values.
The obtained results are organized as follows:
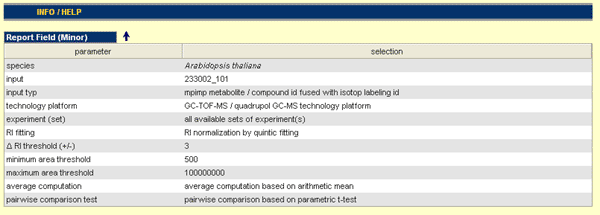
Report Field (Minor)
On the top of the result page you can found the Report Field, which mirrored the parameters used to generate the result set. The result set based on the selected parameters (by user or default) to run this query.
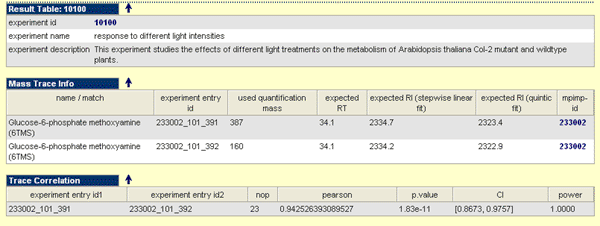
Result Table
The result table covers a short description of the experimental background information. In-depth information can be invoked if you click on the experiment id link:
Mass Trace Info
The mass trace info table covers information regarding to the used mas traces to quantify or qualify the measurement for a particular metabolite. The table is structured as follows:
- Name / Match: The substance name (if mass spectrum identified) of the substance to generate the mass spectrum. If you click on this link a new search with this name will be invoked. If the mass spectrum assigned by match here you can find the details. A matched mass spectrum is represented square brackets with first the match values followed by the substance name / synonym. If you click on this link a new search with this name will be invoked.
- Experiment Entry ID: Is the mass spectral experiment entry id which is fused id (mpimpid_itrid_mass).
- Used Quantification Mass: Mass used for quantification.
- expected RT: Expected retention time (RT) for the fragment.
- expected RI: Expected retention time index (RI) for the fragment according the available regression methods.
- mpimp-id: MPIMP-ID for the substance from which spectrum obtained. If you click on the link you get all access to all entries for this substance.
Trace Correlation
The trace correlation table contains information related to mass trace correlation among the entire replicate set. It provide information to the pairwise comparison of the ids, the number of pairwise observations (nop), the obtained correlation value (e.g. pearson), the probability (p.value), the confidence interval (CI), and the power of test (power).
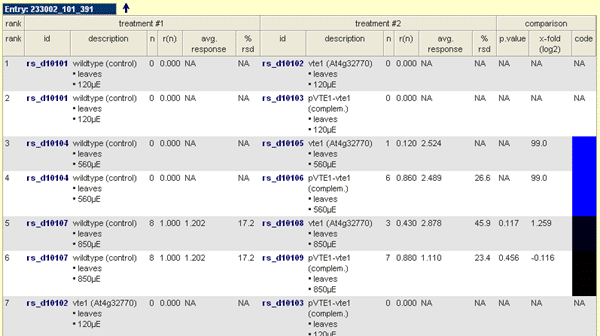
Result Table for Experiment Entry ID
The table is structured as follows:
- Rank: Reflect the original ordering of the pair-wise comparison
- ID: Replicate Set ID for a particular replicate set (set of technical and / or biological replicate measurements [single profiles]). In-depth information can be invoked if you click on the link.
- Description: A short description of the replicate set.
- n: Number of single profiles used for statistical analyses (valid measurements according input parameters).
- r(n): relative number of valid measurements used for statistical analyses out of the set of replicates
- Avg. Response The average response is the mean or median (according user selection) of the replicate set computed for the number of observation from single profiles which fit to the user parameters (valid measurements).
- % r(s/m)d: This value describes the relative standard deviation (if mean selected) or relative median deviation (if median selected) of the replicate set computed for the number of observation from single profiles which fit to the user parameters (valid measurements).
- p.value: The p.value is the observed probability for the pairwise comparison. The computation is done according user selection (parametric or non-parametric statistic selectable).
- x-fold (log2): The x-fold (log2) reflects the increase or decrease level of a particular selected compound normally among the treatment (treatment #1) and the control (treatment #1) sample. The value is log base 2 transformed.
- code: The code is simply a dynamically colour code of the observed x-folds. It is in range from blue to red. A blue colour mirrored increased levels in the treatment (treatment #2). In contrast, a red colour reflects increased level in the control (treatment #1). Black marked cells represent an unchanged value. If you observed NAs no valid measurements are available. If a NA observed for one treatment the artificial values of -99 or 99 (depending if treatment or controld) are used to compute the x-fold.